On August 31,
This FDA guidance is applicable to all devices as defined under section 201(h) of the Federal Food, Drug, and Cosmetic Act. However, this guidance does not apply to the use of non-clinical data, adverse events reports, secondary use of clinical trial data, or systemic literature reviews.
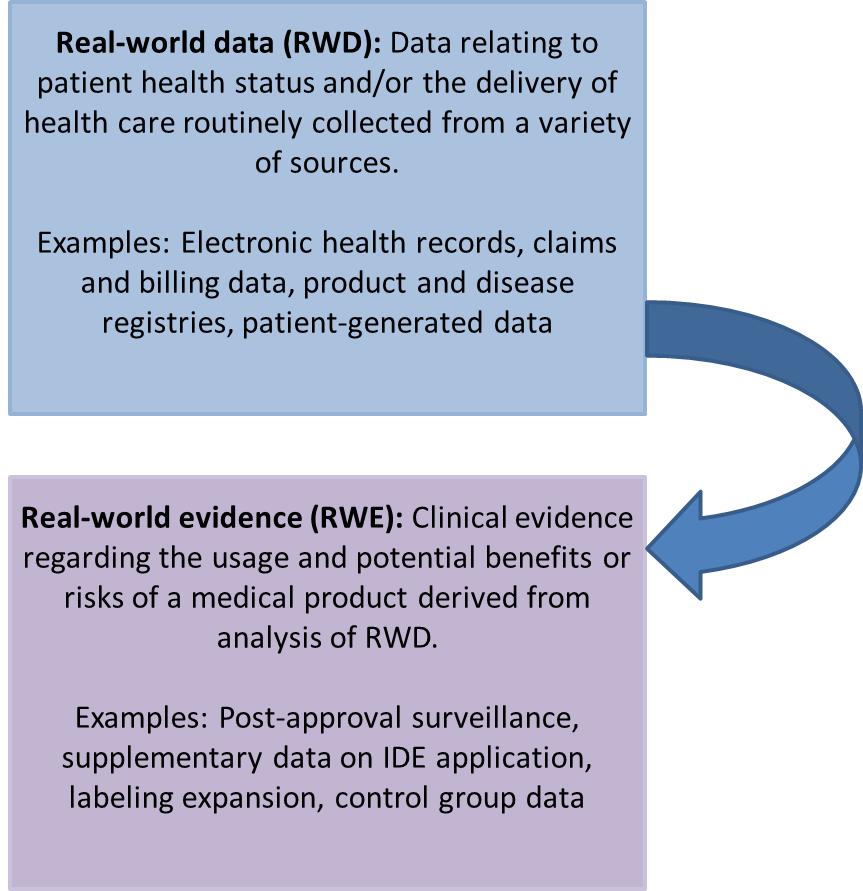
RWD has the potential to facilitate FDA regulatory decisions in the interest of promoting public health and technological advancements while still protecting human safety. During the lifecycle of a medical device, there is a substantial amount of data that is collected over the course of patient treatment and management outside the scope of a clinical trial setting. Furthermore, it is quite common for devices to be used in a clinical setting for indications outside the approved use. As such, a large amount of data is generated throughout the lifecycle of a medical device that can impact patient outcomes.
The guidance is as the name implies; the FDA does not mandate a certain type of RWD nor does it specify a preferred type of RWD. The characteristics, type, and relevance of the RWD will be evaluated case-by-case. Thus, it is up to the sponsor to determine what type(s) of RWD is most applicable and should select appropriate RWD sources to address their specific regulatory questions. It is the responsibility of the FDA to deem the RWD collected to support RWE of sufficient quality for regulatory use.
Do you have experience with the collection or analysis of RWD to support RWE? Has this led to a regulatory decision by FDA?