
Medical devices are part of a multi-billion dollar industry that continues to rapidly grow due to technological advances. As a consequence, medical devices have been a hot topic in the mainstream media with coverage in the documentary “The Bleeding Edge” on Netflix and a piece by John Oliver on HBO’s “Last Week Tonight”. In these pieces, the shortcomings of the medical device regulation process in the United States are the focus. And this has sparked some great discussion among medical device clinical professionals and the public alike. However, understanding the medical device regulation process is paramount in order to strive for continuous improvement, updated regulations, and being informed both as a clinical professional and as a patient.
In the United States, it takes approximately three to seven years on average for a new medical device to come to the market. Compared to pharmaceutical drugs, which take on average 12 years, the medical device approval process is significantly faster and less costly. However, compared to our counterparts in the European Union, a device on average that took five years to come to market in the United States only took 11 months in the EU. In light of this information, there is still much to be done to analyze the safety outcomes of the pathways implemented by both the US and EU to bring devices to market.
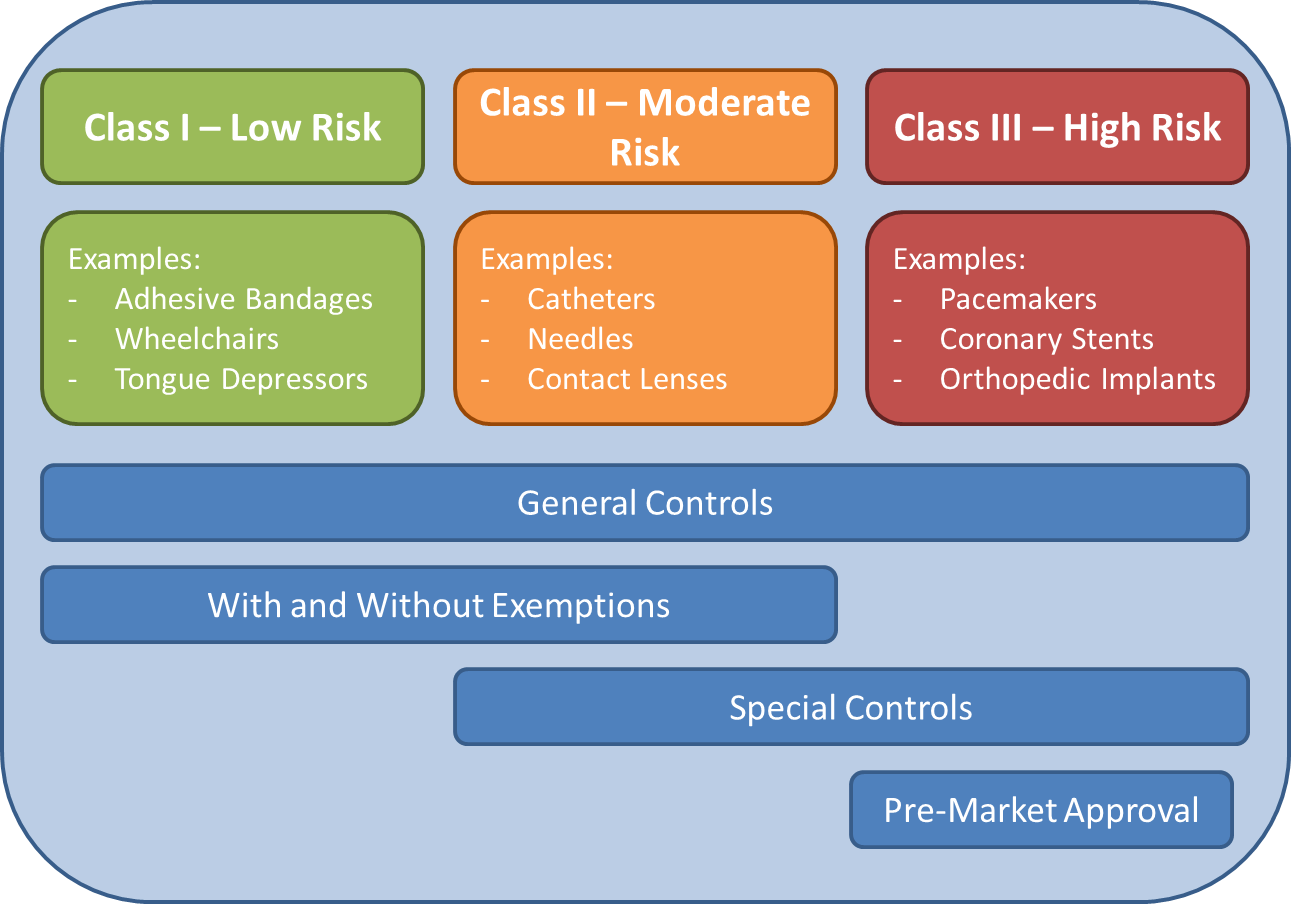
To understand how new medical devices come to market in the United States, we have to start with how medical devices are classified into three categories: Class I, Class II, and Class III. Class I devices are considered low-risk devices many of which can be found in every household such as adhesive bandages, cotton swabs, and toothbrushes. Class II devices have moderate risks and include items such as contact lenses, imaging diagnostic devices (ultrasound, X-rays), and pregnancy tests. And lastly, the highest risk devices fall under the Class III umbrella and include life-saving devices such as pacemakers, coronary stents, and cerebellar neurotransmitter implants.
All devices regardless of the class are subject to general controls by the FDA. A majority of low-risk, Class I devices and a minority of moderate risk, Class II devices can apply for FDA exemption. With an exemption, the FDA will control the manufacturing, product labeling, and risk information that is provided for public use of the device. If the device does not have an FDA exemption, special controls are required prior to marketing the device. And lastly, for high-risk devices, pre-market approval (PMA) is required prior to bringing a device to market. In our next blog post, we will dive deeper into the FDA pathways that are utilized to bring a medical device to market in the United States.
What questions do you have about device classification in the United States?




