
If you missed Part One of our medical device series reviewing medical device classification, please check it out here!
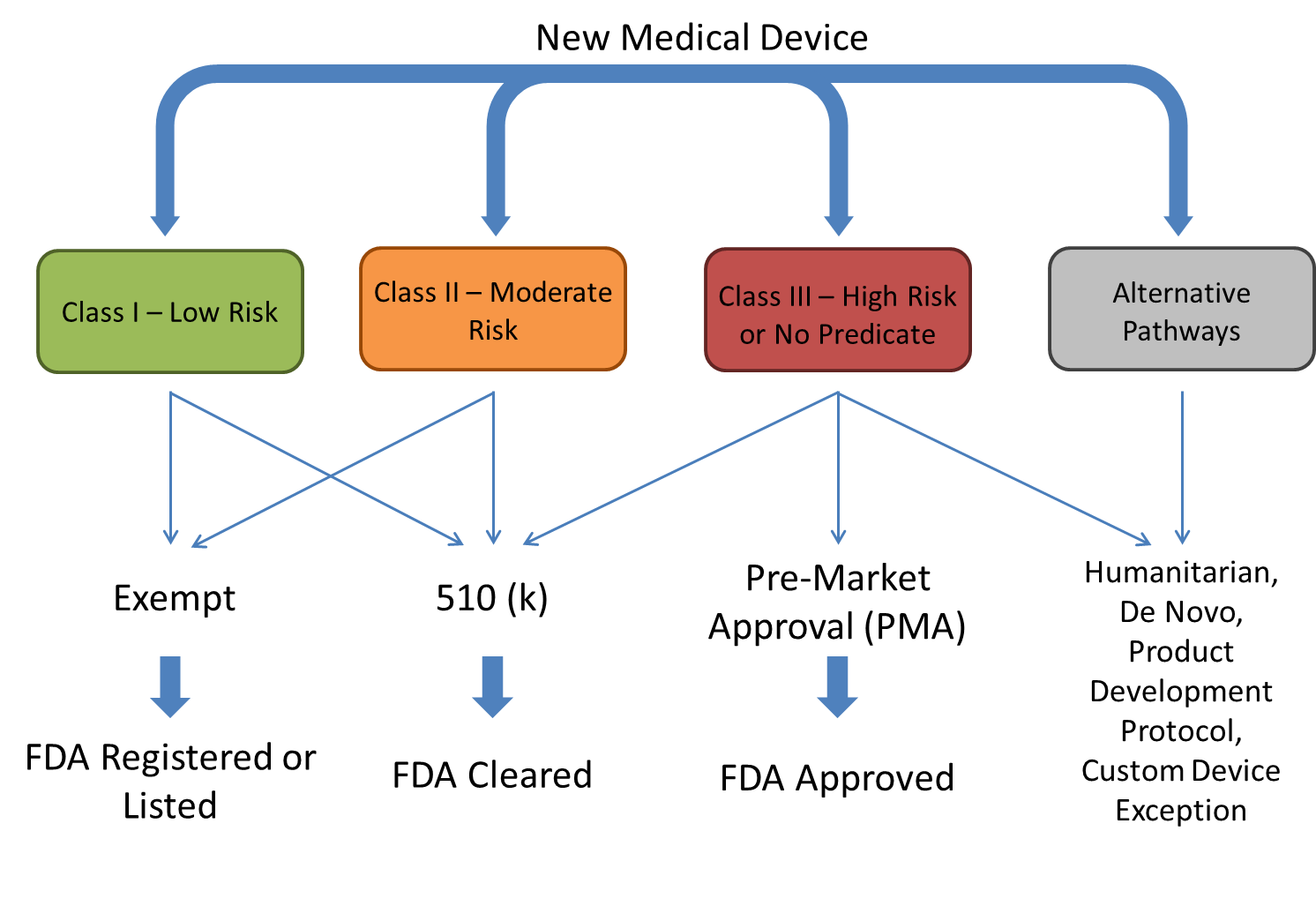
Now that we have a grasp of the FDA medical device classification fundamentals, we can begin to explore the various pathways that a new medical device may follow in order to reach the market in the United States. In most cases, the medical device company will consult with the FDA at the beginning of the process in order to choose the correct pathway to prevent lost time and costs.
Class I and a small amount of Class II devices may apply for exemption with the FDA. If the exemption is granted, no pre-market review is required, but the FDA still controls the labeling and information provided to the consumer. These devices are then referred to as “FDA registered” or “FDA listed” devices once they make it to the market.
If an exemption is not appropriate, the device is low to moderate risk, and there is a device predicate already on the market, the 510(k) pathway is then utilized. A company must prove how the new device is equivalent to the marketed device and provide pre-clinical data, but clinical trial data is usually not required for the 510(k) pathway unless mandated by the FDA. Devices that successfully go through the 510(k) pathway are then referred to as “FDA cleared” devices.
For high risk, Class III devices or for devices that do not have a market equivalent, the pre-market approval (PMA) pathway is required. This requires the company to apply for an Investigational Device Exemption (IDE) from the FDA. Once the IDE is obtained, the company can start the collection of data via a clinical trial in addition to pre-clinical data to provide in the eventual PMA submission. Devices that follow this pathway and are determined safe and effective, receive the “FDA approved” label. For devices that are low to moderate-risk, but do not have predicate on the market, the medical device company can work with the FDA to explore alternative pathways to bring the device to market if clinical trial data is not warranted. Alternative pathways include the De-Novo pathway, Humanitarian Device Exception, Product Development Protocol, and Custom Device Exemption, but these pathways are less common than the 510(k) and PMA pathways.
By the numbers, the vast majority of medical devices are cleared through 510(k) versus approved through the PMA pathway. Approximately 4,000 510(k) applications are submitted to the FDA each year compared to less than 100 PMA applications. The average cost to bring a medical device to market through the 510(k) pathway is $31 million compared to the PMA pathway with average costs of $94 million. The time and cost required to bring a device to market through 510(k) versus PMA makes the 510(k) pathway the popular choice, if available.
Despite the rigorous efforts and controls along the way to bring a medical device to market, the process is not perfect. The push for real-world evidence and post-market surveillance data will continue to shape the medical device landscape in order to ensure patient safety.




