On August 31,
Topics: Medical Devices, New FDA Guidance, Real-World Evidence


You want to keep your trial on track, but physical distancing and travel restrictions have made it difficult. Fortunately, IMARC has experience providing remote services.
On August 31,
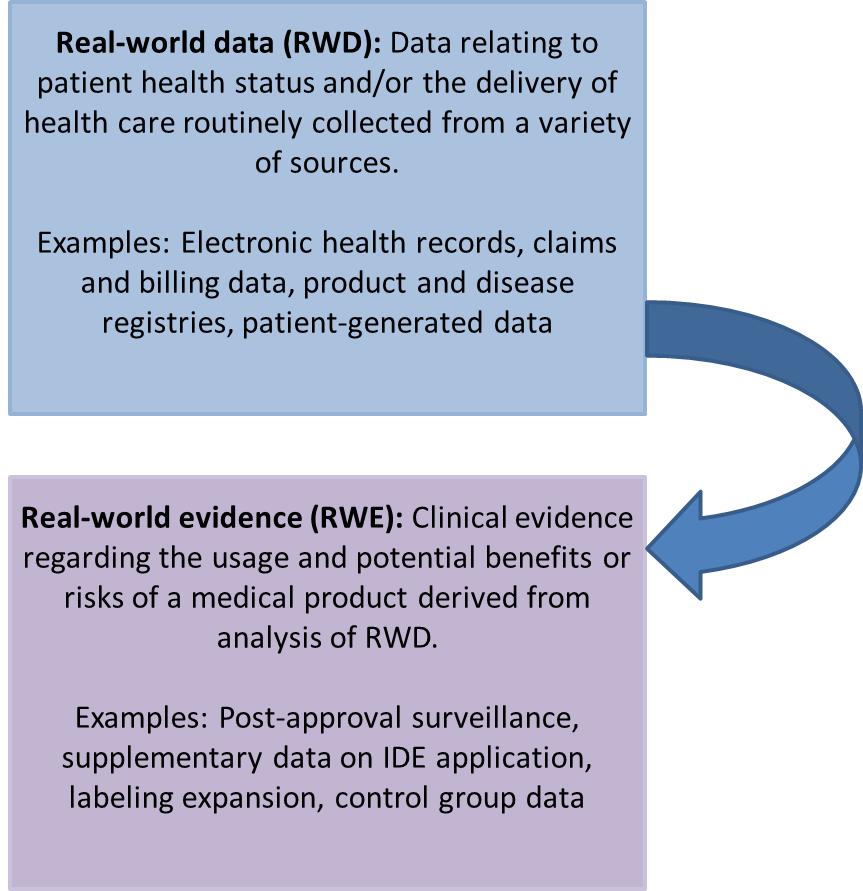
Topics: Medical Devices, New FDA Guidance, Real-World Evidence

In response to The Medical Device User Fee Amendments of 2012 (MDUFA III), which authorizes the FDA to collect user fees for the review of certain premarket submissions, the FDA proposed process improvements to provide further transparency to the review process, including new communication commitments. These new communications are in the context of acceptance review, substantive interactions, and missed MDUFA goals. The communications are outlined in the MDUFA III Commitment Letter and are further described in the draft guidance released on March 5, 2013 titled Types of Communication During the Medical Device Submissions.
Topics: New FDA Guidance, MDUFA, Device Review Process

The FDA has recently released a draft guidance titled “Medical Devices: The Pre-Submission Program and Meetings with FDA Staff” that will update and expand the pre-IDE program established in 1995 and the associated Blue Book Memo from 1999. The program will be renamed the Pre-Submission, or Pre-Sub, program, and will be broadened to include other device submissions, including PMA applications, HDE applications, and 510(k) Submissions.
Topics: New FDA Guidance, Former Pre-IDE Program, Pre-Submission Program
.jpg)
Institutional Review Boards (IRBs), sometimes called Ethic Committees, are responsible for reviewing and approving research studies involving human subjects. The rules that govern IRBs are clearly laid out in 21 CFR 56 which contains the general standards for the composition, operation, and responsibility of an IRB that reviews clinical investigations regulated by the FDA. The IRB has the authority to:
Topics: New FDA Guidance, IRB Oversight, 21 CFR 56, FDA